From CSV to CSA: Implementing Risk-Based Software Validation in Pharma & MedTech
Chapter 1. The problem with Computerized System Validation
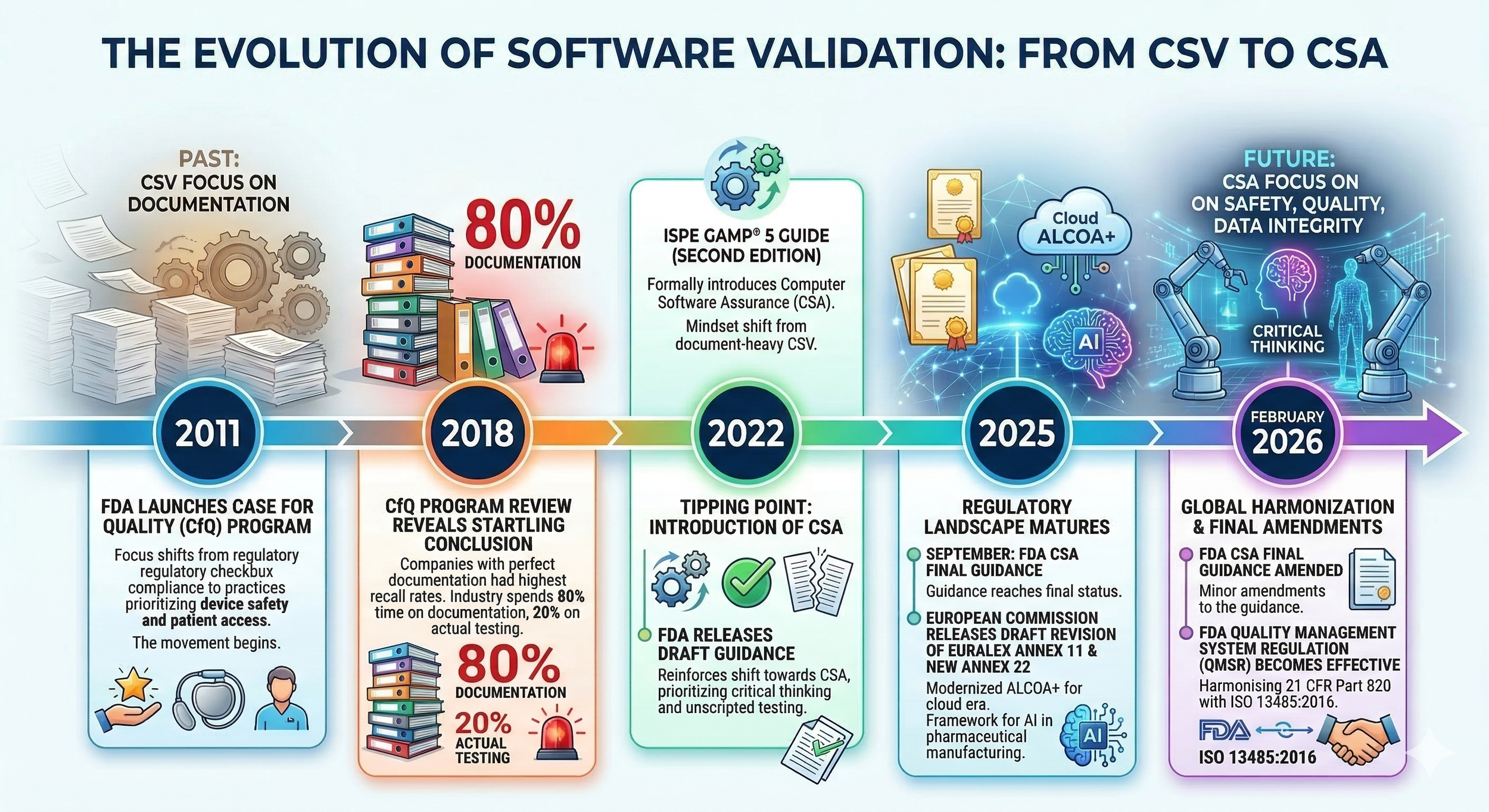
The software validation landscape in the pharmaceutical and medical device industries has shifted rapidly over the last few years. This evolution is driven by the increasing prevalence of digital technologies, including automation, robotics, digital twins, and artificial intelligence. The movement began in 2011 when the FDA launched the Case for Quality (CfQ) program [1]. This initiative encouraged medical device manufacturers to shift their focus from regulatory checkbox compliance to practices that prioritize device safety and patient access. A 2018 review of the CfQ program revealed a fascinating conclusion: companies with perfect documentation often experienced the highest rates of post-market recalls [2]. This led the FDA to a critical realization: the industry was spending 80% of its time on documentation and only 20% on actual testing [3]. This imbalance contradicts the fundamental purpose of compliance, which is ensuring patient safety, product quality, and data integrity.
In 2022, the landscape shifted the release of the ISPE GAMP® 5 Guide (Second Edition) [4]. This revision formally introduced Computer Software Assurance (CSA), which had a fundamental mindset shift away from traditional, document-heavy Computerized System Validation (CSV). CSA demands that we stop treating every software feature in an equal manner. It prioritizes critical thinking and unscripted testing over the industry’s previous focus of exhaustive screenshots and mountains of paper records. The FDA solidified this new way of thinking with its Computer Software Assurance for Production and Quality Management System Software guidance [5]. After years of waiting, it reached Final Guidance status in September 2025 and was further refined in February 2026 to reflect the rapid pace of digital adoption [6].
Simultaneously, in 2025, the European Commission released its draft revision of EudraLex Volume 4, Annex 11 [7], modernizing ALCOA+ requirements for the cloud era. This was joined by the new Annex 22, which provides the first dedicated regulatory framework for Artificial Intelligence in pharmaceutical manufacturing [8]. Annex 11 acknowledges that traditional validation based on CSV simply cannot keep up with self-learning algorithms. Furthermore, as of February 2026, the FDA’s Quality Management System Regulation (QMSR) is officially effective, harmonizing 21 CFR Part 820 with ISO 13485:2016 [9]. Regulators are working together.
The signal from regulators is now clear: they are no longer impressed by the volume of documentation. They are demanding robust, risk-based testing procedures and the application of genuine critical thinking to ensure software actually works as intended. To survive this shift, the industry should move away from the familiarity of CSV and embrace the more rigorous, though less scripted, world of testing.
Chapter 2. Testing what matters: GAMP 5 revision 2 and Software Computer Assurance
What is GAMP?
GAMP is an abbreviation of Good Automated Manufacturing Practice, and it is a framework created by the International Society for Pharmaceutical Engineering (ISPE). The goal is to guide the validation of computerized systems in regulated pharmaceutical and biotech industries. The first edition of GAMP 5 in 2008 introduced the concept of a risk-based lifecycle model, which was further elaborated in revision 2 in 2022. The second edition shifts focus toward patient safety, product quality, critical thinking, and data integrity. It aims to bridge the guideline with modern practices, including cloud and IT service providers, Agile software development, automated tools, data management, and state-of-the-art new technologies, such as Artificial Intelligence and Machine Learning algorithms.
GAMP officially utilizes four distinct categories of software to help scale the validation effort:
Category 1: Infrastructure software (OS, Database engines).
Category 3: Non-configured software (Commercial Off-The-Shelf (COTS) software. This includes typical laboratory GxP systems where the software is used as installed, such as those controlling your FTIR instrument).
Category 4: Configured software (e.g., LIMS, ERP, eQMS).
Category 5: Custom/Bespoke software (High risk, custom code).
As you can see, there is no Category 2. It was removed in earlier versions of GAMP as the industry realized that firmware was better categorized elsewhere. Your CSA activities are supposed to scale depending on the complexity of the category: non-configured software does not require the same elaborate testing as Category 5 software.
To dig a little deeper into the content inside in the second revision:
Critical thinking is emphasized for evaluating risk and tailoring validation approaches. This approach advises companies to question assumptions of what traditional testing activities are necessary, and to utilize vendor-supplied test evidence when appropriate.
Leveraging suppliers and service providers is strongly recommended to avoid duplicative effort in validation. GAMP 5 revision 2 argues that vendors have deep expertise and it should be utilized fully. This makes sense, as companies are moving into the cloud era, where Software-as-a-Service (SaaS) and other service models are becoming increasingly prevalent.
The importance of Agile, iterative development and DevOps for modern software is supported. The guide introduces guidance on how to adopt a sprint-based development model and how to adapt documentation and approval processes accordingly. Principles such as Minimal Viable Products (MVPs) are introduced. The idea is simple: modern software developers have largely been utilizing agile as the development approach for years. It is time for the industry to recognize that waterfall is no longer the only way of developing software for regulated industries.
The use of automation and software tools is emphasized (e.g., eQMS, LIMS). This includes automated controls for risk analysis, change management, and test management. Best modern IT practices are also supported, including continuous integration testing, automated data capture, and other supports that ensure quality and speed.
Validation of emerging technologies is supported. This approach focuses on the model development lifecycle: how to define usage, selecting training data, measuring performance, and monitoring the AI/ML model in production. In 2025, ISPE published a separate GAMP guide on artificial intelligence. If you’re interested, you should check out the GAMP® Guide: Artificial Intelligence [10].
The concept of Computer Software Assurance (CSA) is introduced. The goal is simple: to digitize, automate, and accelerate quality processes to reduce human error. Vendor-supplied validation and targeted, risk-based testing is recommended to reduce excessive and unnecessary documentation.
There are three core foundational pillars in the CSA approach that separate it from traditional CSV:
Critical Thinking: This is the most transformative element of CSA. It requires Subject Matter Experts (SMEs) to engage early in the project to analyze the system's intended use and determine what truly matters to the patient. Critical thinking replaces the static checklist with a scientific inquiry into potential failure modes.
Risk-Based Assurance: CSA moves away from the binary "validated/not validated" state. Instead, it suggests that the rigor of testing and the volume of documentation should be directly proportional to the risk the software poses to product quality or patient safety. Software functions are divided into two categories: High risk and Not high risk.
Leveraging Supplier Evidence: CSA encourages manufacturers to trust the testing already performed by software vendors. If a vendor has a mature quality management system and has already validated a software feature, the manufacturer should leverage that evidence rather than repeating the tests internally. This shifts the focus of the industry toward supplier evaluation rather than redundant testing.
So far in this article, you have seen mentions of scripted and unscripted testing. These practices are elaborated further in GAMP 5:
Scripted Testing: Still required for high-risk functions. This involves a formal protocol with pre-defined steps and expected results. However, even here, CSA suggests reducing documentation. This can be done for example, by capturing a single screenshot of a final state rather than a screenshot for every step.
Unscripted Testing: Used for features that aren’t high-risk. Unscripted testing is performed without a step-by-step script. The documentation only includes the goal, the tester's name, the date, and the final pass/fail conclusion.
Ad-hoc and Exploratory Testing: Used for low-risk features or to probe unexpected system behaviors. This leverages the expertise of the tester to find edge-case failures that a rigid script might miss. This practice makes perfect sense: your CSV expert might have a decade or more worth of experience testing dozens of systems; why limit their expertise by forcing them to follow a script that ignores the bugs they know are likely there?
Next, let’s summarize what we’ve discussed so far by comparing CSV and CSA:

Alright. Now we understand what CSA is and how it compares to CSV. Let’s next look into how you might assess the risk of your software according to GAMP.
Chapter 3. Classifying software according to GAMP 5 revision 2
Building on what we learned in Chapter 2, let’s tailor our approach to a classic QC laboratory example: the Chromatography Data System (CDS). While this software is purchased from a vendor (initially suggesting Category 3), the URS development process often reveals that a CDS is highly configurable. Because of your specific business requirements and the need for custom calculations, the software actually possesses a mixture of Category 3, 4, and 5 features. Fortunately, the majority of the system relies on standard Category 3 COTS capabilities. By utilizing GAMP 5 categories to their fullest, we can assign an overall category of 5 to the system while classifying each feature separately. This allows us to focus our validation activities on the features that actually matter.
The Classification & Risk Assessment Process:
Familiarization: We familiarize ourselves with the software and facilitate a workshop with all necessary stakeholders to list all major features.
Categorization: We classify functions based on GAMP categories: Is it a standard feature (Category 3), a configured feature (Category 4), or a customized calculation (Category 5)?
Impact Assessment: We assess the severity, probability, and detectability of harm, focusing on patient safety and data integrity. Does this function directly impact the data used to release the product? If so, it is high risk.
Risk Leveling: Based on these factors, we assign an overall risk level. High-risk features require robust scripted testing, while low-to-medium risk features may rely on vendor expertise or unscripted testing.
Rationale: We document a rationale for the risk level. This is vital, as it dictates testing complexity. If you intend to reduce testing, your rationale must be robust.
Now, let’s perform an actual risk assessment based on these steps:
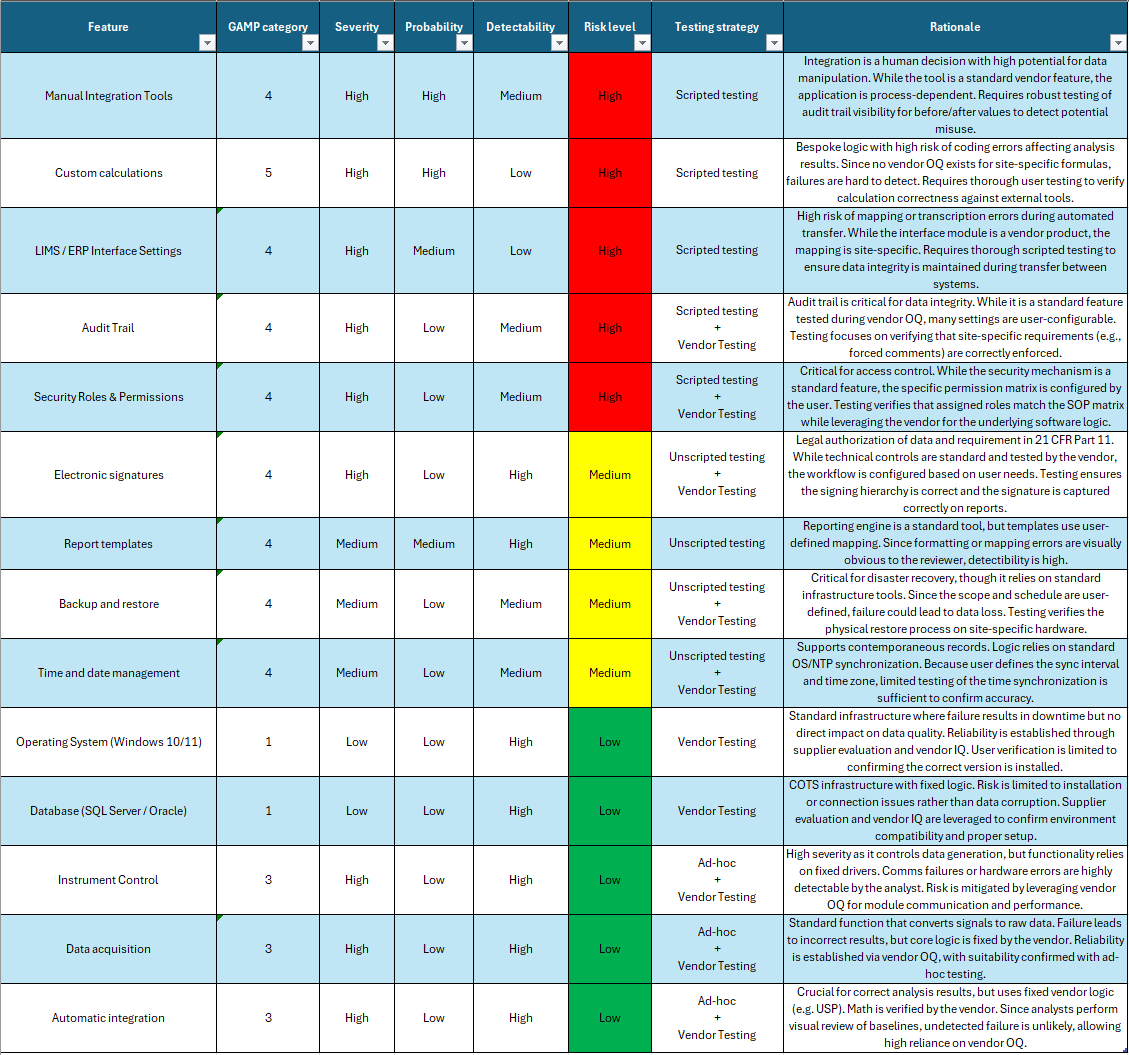
Keep in mind that the results of a risk assessment depend on the specific software and its intended use. The example above may not be suited to your particular quality system, and significant variations are possible. Always tailor your assessment to align with company policies and ensure your rationale is sound.
Chapter 4: The Importance of Change Management
Let’s imagine we have familiarized ourselves with all the regulatory changes related to CSA. We recognize the need for changing our way of working to ensure we are following the modern, best practices. We recognize this change will be gradual and difficult: we cannot simply update our SOPs and switch from CSV to CSA overnight. Like with any changes, this will require rigorious planning, resources and commitment from the entire company.
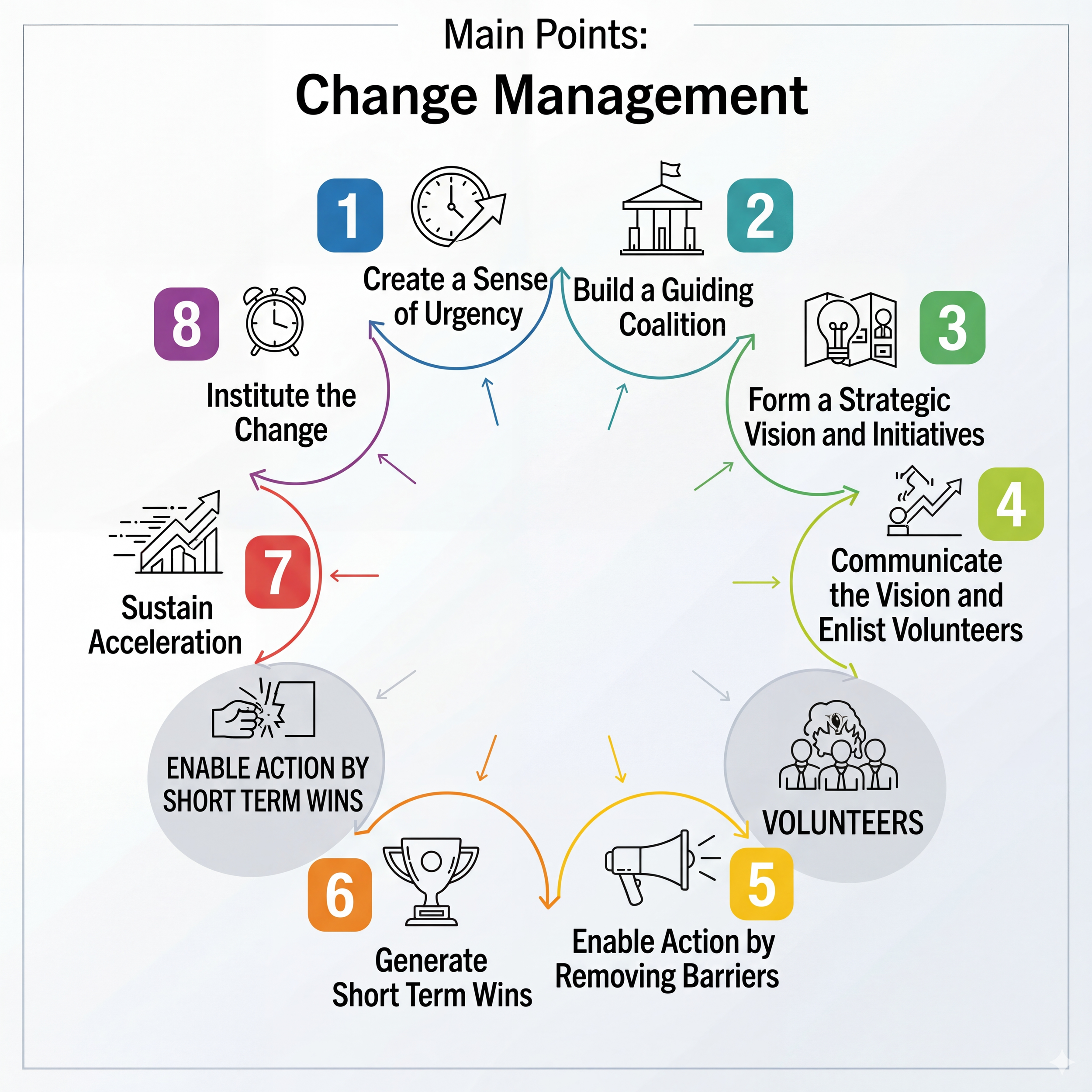
This is why it’s time to talk briefly about change management and design a deliberate change management strategy. For this purpose, let’s adopt Kotter’s 8 step process for leading change [11]:
We create a sense of urgency: We highlight how traditional validation methods create a massive documentation burden that delays the implementation schedules of new GxP systems. By focusing on the risks of stagnant systems, we show that staying with the old CSV model actually increases patient risk rather than reducing it.
We build a guiding coalition: We bring together a cross-functional team of leaders from Quality Assurance, IT and other critical stakeholders to lead the transition. This group ensures that every department feels represented and that no one feels like the new process is being forced upon them without their input.
We form a strategic vision and initiatives: We define a clear path where the goal is value-added validation instead of just checking boxes. Our strategy focuses on applying rigorous testing to high-risk features while simplifying the process for low-risk administrative tasks.
We communicate the vision and enlist volunteers: We share our roadmap with the entire organization to show that the FDA and GAMP 5 guidelines fully support this modern approach. By inviting experienced validation engineers to participate early, we build a group of advocates who are excited to trade paperwork for actual software testing.
We enable action by removing barriers: We update our internal policies and SOPs to remove the requirement for endless screenshots and rigid scripts. This gives our team the formal permission they need to use critical thinking and risk assessments without fearing a negative audit finding.
We generate short term wins: We start with a small pilot project on a low-risk system to prove that the CSA methodology works in a real-world setting. Sharing the data on time saved and bugs found helps convince skeptics that this approach is both faster and more effective.
We sustain acceleration: We take the momentum from our early successes and apply it to more complex GxP systems. We use the time saved on manual documentation to invest in automated testing tools that further improve our software quality.
We institute the change: We make risk-based thinking a permanent part of our company culture by including it in all new hire training and performance goals. Validation is now seen as a continuous process that ensures our software is always safe and effective.
Change is difficult and the complexity and size of your organization will greatly affect the timeline and possibility of instituting a major change like this. Tailor your approach accordingly.
Chapter 5: Reduction of waste with Computer Software Assurance
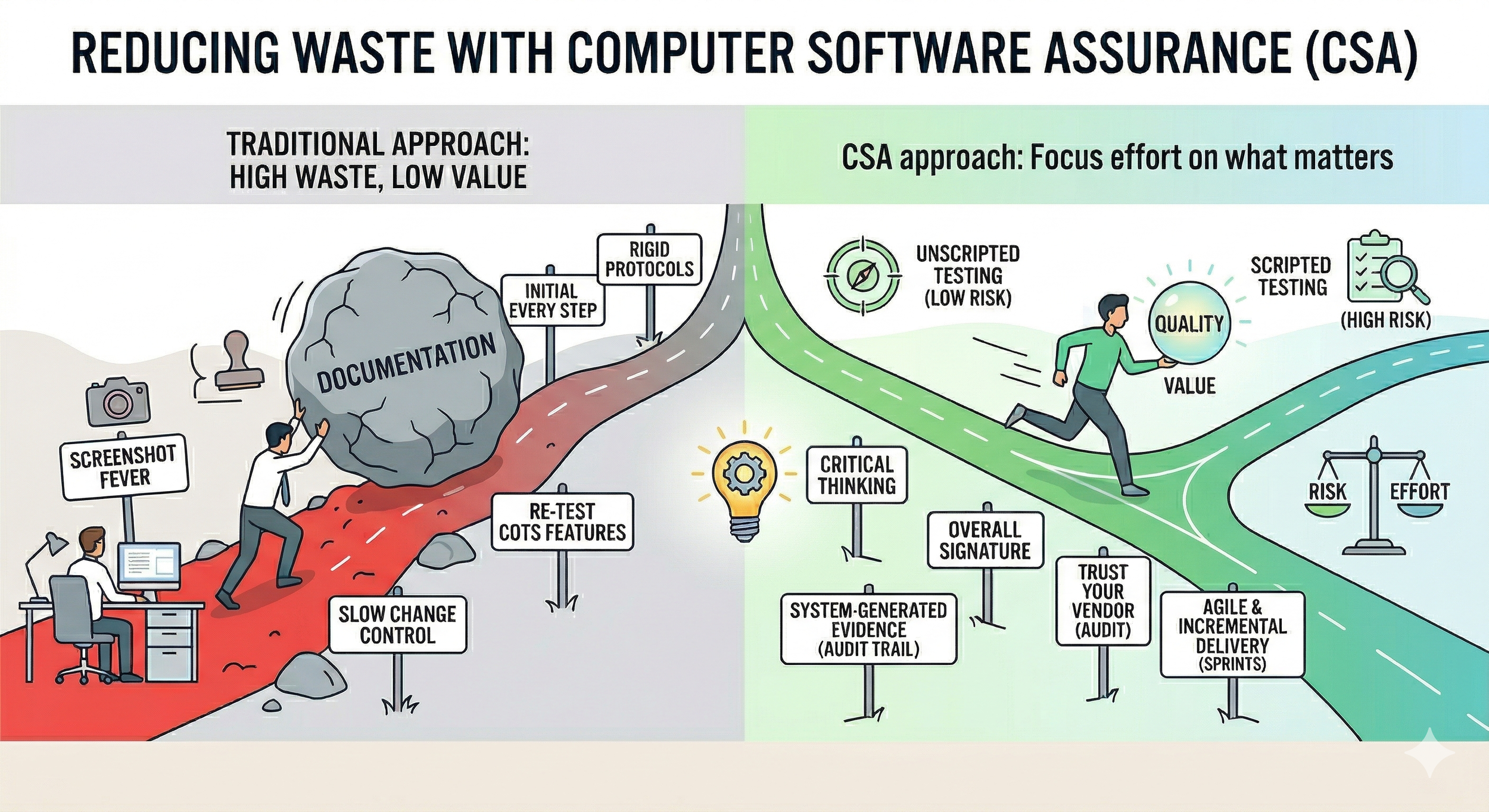
Fundamentally, the transition to CSA is a Lean initiative aimed at creating flow and reducing waste in the software validation process. Regulators agree that the primary goal is to minimize non-value-added activities by focusing on what truly matters. Below are the key strategies for reducing waste:
Critical Thinking: Before outlining specific testing tactics, it is crucial to highlight the driving force behind CSA and GAMP 5 Revision 2: Critical Thinking. By empowering quality and validation teams to apply critical thinking when testing what matters, you give them the regulatory backing to scale their efforts based on actual patient and data risk, granting them permission to say no to unnecessary documentation.
Leverage Unscripted and Ad-Hoc Testing: Use unscripted testing, ad-hoc testing, or vendor test protocols for features that are not high-risk. This significantly reduces the time spent developing detailed protocols, allowing validation engineers to rely on their expertise to explore the system. By providing high-level test objectives rather than step-by-step instructions, testers are encouraged to navigate the software organically. This exploratory approach consistently uncovers real-world bugs that repetitive, inflexible scripts often miss.
Focus Scripted Efforts on High-Risk Functionality: While low-risk features can be verified through exploratory methods, CSA dictates that high-risk functionalities still require scripted validation. For example, if a laboratory uses a LIMS to automatically calculate results critical for batch release decisions, that calculation is a high-risk feature. Your team should focus scripted efforts here, performing negative testing (e.g., inputting out-of-specification data) and stress testing (e.g., simulating massive simultaneous data uploads) to ensure the system doesn't crash or corrupt data.
Use system-generated logs as evidence: Stop requiring screenshots as test evidence for meaningless steps. Under traditional validation, engineers often capture several screenshots for a single workflow just to prove they clicked a button. CSA explicitly discourages this, recommending system-generated logs as objective evidence instead. For example, when verifying a 21 CFR Part 11 electronic signature, do not screenshot every prompt. Instead, execute the workflow and attach a single PDF export of the native audit trail, which provides irrefutable, timestamped evidence.
Reduce unnecessary signing: Reduce the need for manual initials and dates on every single test step. This practice adds significant waste without improving quality. Under the CSA framework, the focus shifts to verifying the overall intent of the feature. If a validation engineer is testing an HPLC sample analysis sequence, they shouldn't be forced to sign off on sub-steps. If the overarching acceptance criteria are met, a single Pass with the tester's signature at the end of the run is sufficient.
Adopt Agile and Incremental Delivery: If your company develops GxP-grade software, ensure your quality system supports Agile practices. Leveraging 2–4 week sprints to develop software incrementally allows you to utilize automated testing for mini-validations between iterations. This helps you fail fast and resolve issues early, rather than discovering that a feature doesn't meet user needs at the end of a two-year project.
Simplified Change Control: Create a simplified change control process for minor software patches or low-risk updates. If you are forced to go through rigid planning for every minor update, you are unlikely to keep your GxP-critical software up to date. Outdated, bugged software does not improve patient safety. Under CSA, the team can execute a rapid impact assessment, confirm there is no risk to data integrity, and deploy the patch swiftly using the vendor’s release notes as evidence.
Trust your vendors: Maximize vendor leverage to eliminate redundant testing, especially for SaaS and cloud applications. Do not waste time re-testing standard COTS features that the supplier has already verified. GAMP 5 Revision 2 emphasizes that if a robust supplier assessment confirms mature quality practices, you should accept their validation documentation (such as their OQ). Your internal efforts can then remain lean, focusing only on custom configurations and intended use.
Sources:
[1] FDA (2024) Case for Quality. [Online] Available at: https://www.fda.gov/medical-devices/quality-and-compliance-medical-devices/case-quality [Accessed 31 March 2026].
[2] Medical Device Innovation Consortium (MDIC) (2022) Case for Quality (CfQ) Pilot Program Summary. [Online] Available at: https://mdic.org/wp-content/uploads/2022/05/CFQ.pdf [Accessed 31 March 2026].
[3] Medical Device Innovation Consortium (MDIC) (2019) CSA: Computer Software Assurance for Manufacturing, Operations, and Quality System Software. [Online] Available at: https://mdic.org/wp-content/uploads/2020/10/2019.04.23-IVT-FDA-Industry-Team-CSA-Presentation-Final.pdf [Accessed 31 March 2026].
[4] ISPE (2022) GAMP 5 Guide: A Risk-Based Approach to Compliant GxP Computerized Systems. 2nd edn. North Bethesda: ISPE. Available at: https://ispe.org/publications/guidance-documents/gamp-5-guide-2nd-edition [Accessed 31 March 2026].
[5] FDA (2022) Computer Software Assurance for Production and Quality System Software: Draft Guidance for Industry and Food and Drug Administration Staff. [Online] Available at: https://www.fda.gov/media/162627/download [Accessed 31 March 2026].
[6] FDA (2025) Computer Software Assurance for Production and Quality Management System Software: Guidance for Industry and Food and Drug Administration Staff. [Online] Available at: https://www.fda.gov/media/188844/download [Accessed 31 March 2026].
[7] European Commission (2025) EudraLex Volume 4: Good Manufacturing Practice (GMP) Guidelines, Annex 11: Computerised Systems (Consultation Document). [Online] Available at: https://health.ec.europa.eu/document/download/40231f18-e564-4043-94de-c031f813d38b_en?filename=mp_vol4_chap4_annex11_consultation_guideline_en.pdf [Accessed 31 March 2026].
[8] European Commission (2025) EudraLex Volume 4: Good Manufacturing Practice (GMP) Guidelines, Annex 22: Artificial Intelligence (Consultation Document). [Online] Available at: https://health.ec.europa.eu/document/download/5f38a92d-bb8e-4264-8898-ea076e926db6_en?filename=mp_vol4_chap4_annex22_consultation_guideline_en.pdf [Accessed 31 March 2026].
[9] FDA (2026) Quality Management System Regulation (QMSR). [Online] Available at: https://www.fda.gov/medical-devices/postmarket-requirements-devices/quality-management-system-regulation-qmsr [Accessed 31 March 2026].
[10] ISPE (2025) ISPE GAMP® Guide: Artificial Intelligence. North Bethesda, MD: International Society for Pharmaceutical Engineering. Available at: https://ispe.org/publications/guidance-documents/gamp-guide-artificial-intelligence(Accessed: 4 April 2026)
[11] McCalman, J., Paton, R.A. and Siebert, S. (2016) Change management: a guide to effective implementation. 4th edn. London: SAGE Publications